
Research
General Outline
We are engaged in theoretical and computational investigations of complex chemical and biological systems in solution with atomic detail. Besides molecular dynamics (MD) simulations and dielectric continuum models in conjunction with quantum chemistry, we focus methodically on liquid state theory for modeling solvation effects. Our characteristic conceptual platform is integral equation theory in various flavors. Numerical solutions to these theories yield approximate distribution functions, for instance for solvent particles around solute molecules or near interfaces. From these data we can compute various structural and thermodynamic properties. The methods are specifically designed to work in conjunction with quantum chemistry (to account for electronic response upon solvation) and molecular dynamics (MD) simulations (to consider conformational flexibility of species in solution). Applications range from organic chemistry in aqueous and nonaqueous solution up to problems in biophysics and drug development.

Topics
Conceptual and numerical developments for integral equation theories
We try to systematically understand and improve the approximations involved with integral equation theories as well as the numerical difficulties associated with their numerical solution on large three-dimensional grids. In particular, we ask for fundamental relations to derive the free energy of liquids and solvated species from knowledge of the solute-solvent interactions described at the same level of detail as employed in MD simulations. A range of exact results have been obtained in the past to allow for further developing the methods with the ultimate goal to reach quantitative agreement with MD results at a fraction of computational cost.
Quantum chemistry in solution and in heterogeneous systems
Electronic structure calculations are combined with integral equation theory by our “embedded cluster reference interaction site model” (EC-RISM) in order to arrive at a tractable and predictive theory for solvent and environmental effects on chemical reactivity. Briefly, electronic and liquid structure are computed self-consistently by mapping the solvent charge distribution onto discrete point charges which polarize the electronic Hamiltonian. We obtain the solvent distribution and associated correlation functions to deduce the solvation free energy along with a molecule’s wave function in solution, from which electronic properties can be derived. Typical applications (to be detailed below) range from predictions of aqueous and nonaqueous acidities (pKa) and tautomer equilibria in homogeneous solution or near interfaces up to estimating the electronic response of solute species exposed to extreme thermodynamic conditions.
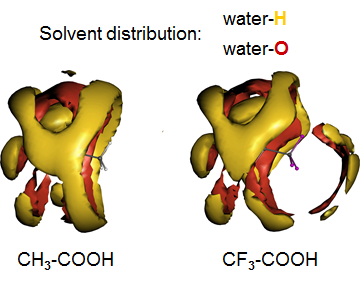
Chemistry and physicochemical properties of small drug-like molecules
A key problem in medicinal chemistry and drug development is related to predicting physicochemical properties such as acidity constants and distribution coefficients between aqueous and nonaqueous phases, coupled to the chemical problem of characterizing the relevance or population of certain tautomeric and conformational states. To this end, EC-RISM calculations can be employed, assessing the quality of which require benchmarking against experimental data. A statistically sound measure of prediction quality is provided by blind prediction contests such as the SAMPL series. Our latest contributions to these challenges involve tautomer abundance prediction and pH-dependent distribution coefficient calculations (log D) between aqueous and nonaqueous phases.
Spectroscopic properties of solvated species
The availability of a solvent-modulated electronic wave function of a solute molecule from EC-RISM calculations allows for the determination of wave function-derived spectroscopic features. We specifically target NMR parameter prediction in solution under varying conditions in order to determine the impact of the environment. This way, it is possible to distinguish conformational from solvation effects such that structure determination in solution becomes more reliable. Solvation effects on vibrational modes to be studied by IR spectroscopy are also an important area of research, where the challenge is to account for nonequilibrium effects from a theory of the equilibrium state.
Chemistry at extreme conditions
Extreme thermodynamic conditions such as high hydrostatic pressure are parameters to modulate chemical and biological systems in a way not possible under ambient temperature and pressure. Studying systems at extreme conditions is furthermore relevant for understanding the essential features of life-forming compounds and their adaptive capabilities. We developed special EC-RISM techniques in collaboration with theoretical and experimental groups that allow for reliable calculations of electronic structure at high pressure. From these data it is possible to derive high pressure-optimized force fields for further MD simulations and for studying the impact of extreme conditions on spectroscopic features.
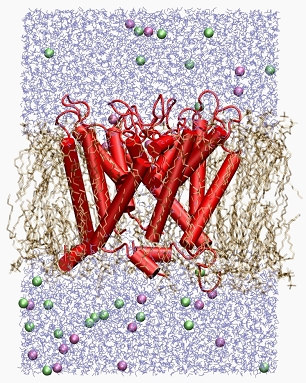
Ion channel function
Channel proteins, embedded in lipid bilayers, are studied by extensive MD simulations in combination with integral equation analyses of ion distributions in order to identify correlations between sequence and conductance/selectivity properties. A particularly difficult question is how and why the protein switches between conformational states of varying conductance ("gating"). Typical simulation systems contain on the order of 105 atoms and are computed over several hundreds of nanoseconds. The combination of MD and RISM theory allows for tackling the combined problem of conformational variability and preference for certain ion species. We apply the methodology for instance to potassium channels such as the miniature channel protein Kcv and derivatives that are experimentally studied by G. Thiel at TU Darmstadt/Germany.
Protein-ligand binding
Integral equation calculations for the solvent degrees of freedom are applied in conjunction with MD simulations for proteins and ligands. This model is currently developed for drug development purposes in collaboration with industrial partners via affinity prediction, binding pose ranking and local hydration analysis. Results are benchmarked against experimental data provided by collaboration partners and against explicit-solvent free energy simulations. More specifically, the key question of “displaceable water” sites that can be targeted by a ligand can be addressed by an integral equation approach without suffering from sampling problems frequently encountered with MD for low-occupancy regions. A recent development, “binding free energy derivatives”, allows for defining a search direction in “chemical parameter space” that aims at rationally finding ligand chemistry variations that optimize the binding affinity.