
Projects
The following sections highlight achievements of the Clever Lab and ongoing research projects.
Cage Assembly, Ligand Functionalization and Host-Guest Studies
Standing on big shoulders, our lab’s journey into the area of self-assembled coordination cages began with the study of a family of interpenetrated double cages, each consisting of a pair of catenated [Pd2L4] lantern-shaped helicates (Frank CEJ 2016; Han CSR 2014). In contrast to monomeric cages, featuring one large cavity to bind guest molecules, the mechanically interlocked dimers have three pockets to bind small guests. We could show that anion encapsulation in the outer two binding sites proceeds following an allosteric mechanism with positive cooperativity (Freye ACIE 2012, JACS 2013). The exchange of larger anions for smaller ones in the outer two pockets leads to a relative motion of both [Pd2L4] along the common Pd4-axis, thereby enlarging the central cavity (Freye CEJ 2013; Dieterich PCCP 2012). In further work, we showed that this can be used to trigger recognition of small uncharged guest molecules in this cavity, largely driven by London dispersion interactions (Löffler JACS 2015, Chem. Commun. 2017).

Figure 1: a) Metal-mediated self-assembly of banana-shaped ligands to a Pd2Ligand4 coordination cage and dimerization to an interpenetrated double cage; b) allosteric binding of chloride in the two outer pockets triggers uptake of neutral guests in the central cavity.
Our interest in assemblies showing intricate shapes and topologies led to the synthesis and characterization of a series of further cages with peculiar structure. Some examples are shown in the following figure.

Figure 2: Examples for self-assemblies with reduced symmetry and intricate topology: a) cuboid box-shaped cage (Han CEJ 2014); b) interpenetrated dimer of peanut-shaped cages (Zhu ACIE 2018); c) huge Pd8L16 catenane (Bloch ACIE 2018); d) double-trefoil knot (Engelhard ACIE 2012).
In parallel, we started to equip monomeric and interpenetrated cages with functional backbones. In one study, we compared the influence of inside pointing (“endohedral”) substituents on cage dynamics and guest binding, showing that [Pd2L4] cages with four bulky adamantyl groups show pronounced flipping motion and allow guest binding in an adaptive manner (Löffler Chem. Sci. 2016). In a related study, we showed that the spinning of rotors pointing inside the cage cavity are modulated by their electronic push-pull character and the presence of guests (Krick Chem. Commun. 2016).

Figure 3: Cages with pronounced endohedral dynamics, modulated by guest binding: a) flipping of bulky ligands; b) rotation of rotors around push-pull-activated double bonds.
In another approach, we started to equip cages with chromophore-based organic backbones to exploit the interaction of supramolecular structures with light. By using bis-monodentate ligands based on dithienylethene (DTE) photoswitches, we could demonstrate light-triggered guest uptake and release as well as reorganization of small rings into large spheres (Han ACIE 2013, ACIE 2016; Li JACS 2019). In a system combining electron-rich donor and electron-poor acceptor functionality within an interpenetrated double cage, we were able to identify light-powered charge separation via time-resolved UV-Vis and IR spectroscopic investigations (Frank ACIE 2013, JACS 2016). A drawback of this system, however, was its statistical combination of donor and acceptor moieties, without control over stoichiometry and stereochemistry of the mixture of related assemblies. This problem turned into one of our major motivations to develop strategies towards non-statistical assembly of coordination structures, as detailed in the following section.

Figure 4: Light-triggered processes in stimuli-responsive cages: a) photoisomerization of a DTE based Pd2L4 cage between a guest-binding host and its guest-releasing isomer; b) reversible structural conversion between small rings and large spheres by irradiation with light of different wavelengths.

Figure 5: Donor- and acceptor-functionalized cages dimerize to mixed-ligand interpenetrated structures with closely packed electron-rich and electron-poor chromophores that show light-powered charge separation.
Non-Statistical Assembly Strategies for Multifunctional Cages
We develop a number of orthogonal approaches to allow the targeted formation of self-assembled structures under thermodynamic control from more than one type of ligand without running into statistical mixtures (or narcissistic self-sorting). The following figure gives an overview of assembly strategies towards heteroleptic coordination cages currently under development in the Clever Lab (Bloch Chem. Commun. 2017; Pullen Acc. Chem. Res. 2018).

Figure 6: Schematic representation of self-assembly strategies leading to the non-statistical formation of mixed-ligand coordination cages.
Structures formed by such “integrated self-sorting” strategies can be equipped with more than one kind of ligand-centered functionality. We study the interplay of these different chemical modifications, leading to emergent system behavior, control over processes inside the cage cavity and interesting material properties (Saha JACS 2018). One aspect deals with the interaction between light-harvesting and redox-active components. Here, we study exciton and electron transfer between host and guest structures (host-to-guest or guest-to-host), within heteroleptic assemblies (ligand-to-ligand) or individual supramolecules in more complex systems. While such processes are relevant from a fundamental energy conversion viewpoint, we regard these effects also in terms of propagating information on a molecular scale. As such, we also study chirality transfer within host-guest systems and mixtures of interacting supramolecular assemblies (Li JACS 2019).

Figure 7: Examples for heteroleptic cages formed by the coordination-sphere engineering approach (left; Zhu CEJ 2018) and the shape-complementarity principle (right; Block JACS 2016, ACIE 2017).
Selective Recognition, Confined Catalysis and Adaptive System Behavior
With numerous cage compounds of different size, shape and functionalization in hand, we utilize these compounds in the context of selective recognition, reactions under confinement and the realization of interacting networks. For example, we could show that moving from homoleptic [Pd2L4] cages, featuring coplanar Pd(pyridine)4 units connected by four banana-shaped ligands of the same chemical structure, to heteroleptic cis-[Pd2L2L’2] cages, having a curved shape, guest binding preference goes from linear to bent molecules (Bloch JACS 2016). Homochiral cages based on ligands with a helicene backbone were shown to undergo chiral self-sorting and recognize guest enantiomers (Schulte ACIE 2019).

Figure 8: Comparison of a) cavity morphologies and b) suitable guest geometries realized by different assembly strategies.
By using bowed ligands presenting their π-surfaces into the cavity of self-assembled cages and bowls, we were able to obtain new fullerene-binding host systems that alter their guests’ electronic situation, make them soluble in a range of polar organic solvents and act as supramolecular protecting groups to steer covalent fullerene modification (Chen JACS 2019, CEJ 2019). In another line of research, we employ chromophore-functionalized cages to develop light-triggered and photo-redox reactive and catalytic systems. Using acridone-based interpenetrated double cages, we could show O2 sensitization, followed by hetero-Diels-Alder addition chemistry (Pullen Dalton Trans. 2020). Both the substrate as well as product were characterized in form of their host-guest complexes with the self-assembled cage. We have recently extended such studies to a series of further dyes. In addition, we study the effect of photoswitchable chromophores in the context of adaptive and non-equilibrium systems of interacting supramolecules.

Figure 9: Control of chemical reactions with self-assembled coordination cages: a) Quinoline-based ligands lead to assembly of Pd2L3 bowls, binding fullerenes and allowing their selective mono-functionalization; b) an acridone-based double cage acts as a chemically robust singlet oxygen photosensitizer.
The Clever Lab has a special expertise in the chiroptical characterization of coordination compounds and supramolecular systems, running an Applied Photophysics Chirascan CD spectrometer and a JASCO CPL-300 spectrometer to record circularily polarized luminescence. Examples from our recent work include the use of chirality transfer to elucidate the temporal course of guest localization inside/outside the cavity of a light-switchable cage during a stepwise photoswitching sequence (Li JACS 2019). We further recorded the CD-spectroscopic response of the guest-induced compression/elongation of chiral cages consisting of four spring-shaped helicenes to differentiate non-chiral guests by chiroptical methods (Schulte ACIE 2019). In the context of organometallic chemistry, a unique chiral-at-metal, luminescent square-planar platinum(II) compound composed of achiral, trans-chelating ligands was synthesized, optically resolved and characterized by CPL spectroscopy (Schulte JACS 2017).

Figure 10: Chirality transfer from guest to the host structure of a photoswitchable cage allowed us to determine the light-triggered mechanism of the cage release mechanism.
A further technique that has added significant momentum to understanding size, shape and dynamics of our heteroleptic cages and host-guest complexes and helps us analyzing complex mixtures is ion mobility mass spectrometry, performed on a Bruker timsTOF instrument. This technique allows us to differentiate species in complex mixtures that NMR (including DOSY) techniques cannot resolve and HPLC cannot perform for reasons of compound stability (Ebbert Dalton Trans. 2019; Schulte ACIE 2020). Furthermore, we elaborate a number of theoretical workflows for the calculation of collisional cross section values to assign experimental ion mobilities to structural models of isomeric species. Examples are shown in the following figure.

Figure 11: Chiroptical effects in coordination compounds: a) binding of guests of different lengths leads to CD- and ion-mobility spectrometry readout in chiral helicene-based cages; b) unique chiral-at-metal luminophors based on square-planar platinum(II) complexes with trans-chelating ligands showing circularly polarized luminescence.
Heteroleptic Transition Metal Environments in DNA G-Quadruplexes
The concept of metal-mediated base pairing in the context of double-stranded DNA, where the exchange of canonical nucleobases by ligands allows for the incorporation of defined transition metal complexes inside nucleic acids, has extensively been studied in the last decades. Although quite elaborate systems could be realized with duplex DNA, reports about the expansion of this concept in other DNA secondary structures remain scarce. In the Clever Lab, we recently introduced a robust and synthetically feasible approach to covalently incorporate ligands such as pyridines or imidazoles into DNA G-quadruplex-forming sequences. Folding of these ligand-modified oligonucleotide sequences into defined tetramolecular stacks of guanine-quartets leads to a pre-arranged ligand environment suitable for the reversible binding of transition metal ions like Cu2+, Ni2+ or Zn2+ (Figure 12; Engelhard ACIE 2013, CEJ 2018; Punt Chem. Sci. 2019). A substantial thermal stabilization of the metal ion-bound secondary structures has been observed, which could be probed by UV/Vis- and CD-based melting experiments.

Figure 12: Formation of metal-bound G-quadruplex DNA from ligand-modified guanine-rich oligonucleotides by hybridization in aqueous buffer and chelation of a transition metal ion (here Cu(II)).
Based on telomeric or other non-coding sequences derived from the genome of different organisms, we obtain unimolecular quadruplexes folded from a single strand, with loops spanning over sides and faces of the G-quartet stacks. By synthetic incorporation of metal binding sites close to or in the loop regions, we demonstrated stimuli-responsive functionality, e.g. copper-induced switching between different unimolecular folding topologies (Figure 13). Further, the known DNA-protein interaction between a specific quadruplex sequence (tba, thrombin binding aptamer) and thrombin could be controlled in a metal-dependent way, as monitored by a fibrinogen clotting assay (Engelhard ACIE 2017).

Figure 13: Copper-induced switching between different G-quadruplex folding topologies.
We further demonstrated that variation of ligand number and position in unimolecular G‑quadruplexes enables a controlled design of distinct coordination environments with fine-tuned metal affinities (Punt CEJ 2019). We use these metal-carrying G‑quadruplexes in enantioselective catalysis (Figure 14). Their straightforward synthetic accessibility and well-defined coordination environments allow fine-tuning of catalytic activity, even leading to switchable enantioselectivity (Punt JACS 2021). Based on this results, we are interested in expanding the scope of ligand derivatives since the single-oligonucleotide nature of the folded telomer sequences allows us to mix different ligands in the pocket created by the loops, thereby opening the potential to mimick metallo-protein activity inside an oligonucleotide environment. We approached this concept combining different ligands in G-quadruplex-forming sequences creating distinct heteroleptic coordination environments (Punt Front. Chem. 2020).
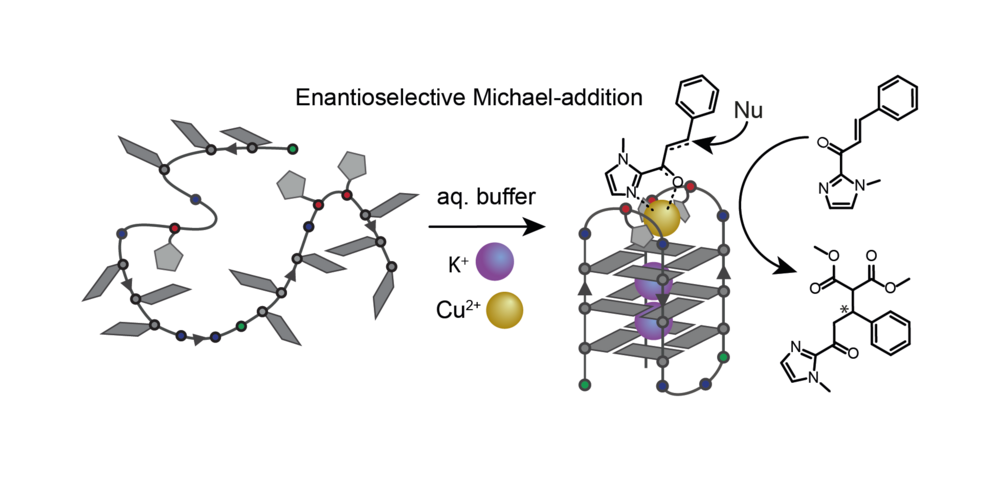
Figure 14: Exemplary enantioselective catalytic conversion in the metal-carrying loop region of a unimolecular G-quadruplex.
Furthermore, we exploited the paramagnetic nature of bound Cu2+ ions in G-quadruplex structures to design new probes for EPR-spectroscopic distance measurements on the nano scale. In the first step, intramolecular Cu2+-Cu2+ distances were quantified with the help of pulsed EPR PELDOR and RIDME measurements in tetramolecular structures bearing metal pyridine tetrads on both faces of the guanine stack, serving as a proof-of-principle EPR-based ruler (Engelhard Chem. Commun. 2018). This concept has then been extended to investigate intermolecular distances in higher-order G‑quadruplex structures like dimers and sandwich complexes (Figure 15), thus establishing a new, highly precise tool for DNA structure elucidation (Stratmann ACIE 2020).

Figure 15: EPR distance determination in higher-order G-quadruplex structures such as dimers and sandwich complexes with quadruplex-binding molecules. A molecular dynamics-derived structural model in full accordance with the EPR data is shown on the right.